Artículo de Revisión
Coronary thrombus: pathophysiology and consequences
Sundararajan Srikanth, John Ambrose
Revista Argentina de Cardioangiología Intervencionista 2012;(01): 0009-0017 | Doi: 10.30567/RACI/201201/0009-0017
Atherosclerosis is a systemic vascular pathology that is preceded by endothelial dysfunction. Vascular infl ammation promotes atherosclerosis and creates the milieu for episodes of intravascular thromboses. Thrombotic events in the coronary vasculature may lead to asymptomatic progression of atherosclerosis or could manifest as acute coronary syndromes or even sudden coronary death. Thrombus encountered in the setting of acute coronary syndromes has been correlated with acute complications during percutaneous coronary interventions such as no-refl ow, acute coronary occlusion and long term complications such as stent thrombus. This article reviews the pathophysiology of coronary thrombogenesis and explores the complications associated with thrombus during coronary interventions.
Palabras clave: coronary thrombus, percutaneous intervention, endothelial dysfunction, atherosclerosis.
La aterosclerosis es una enfermedad vascular sistémica que es precedida de la disfunción del endotelio de los vasos arteriales. La inflamación vascular promueve la enfermedad aterosclerótica y es la base para el desarrollo de trombosis intravascular.
Los eventos trombóticos en el árbol arterial coronario pueden conducir a la progresión asintomática de la aterosclerosis coronaria o manifestarse también como un síndrome coronario agudo y/o episodios de muerte súbita. Estos episodios de trombosis coronaria durante los síndromes coronarios agudos también se pueden correlacionar con las complicaciones trombóticas registradas durante procedimientos de angioplastia coronaria como: el no-reflujo, la oclusión coronaria aguda y la trombosis del stent tanto temprana como tardía.
Este artículo revisa las diferentes fisiopatologías de la trombosis coronaria, las complicaciones trombóticas de los procedimientos percutáneos coronarios así como su posible prevención y tratamiento.
Keywords: trombo coronario, intervención percutánea, disfunción endotelial, aterosclerosis.
Los autores declaran no poseer conflictos de intereses.
Fuente de información Colegio Argentino de Cardioangiólogos Intervencionistas. Para solicitudes de reimpresión a Revista Argentina de Cardioangiología intervencionista hacer click aquí.
Recibido 2012-05-02 | Aceptado 2012-05-05 | Publicado

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-SinDerivar 4.0 Internacional.







Introduction
Extensive research in vascular biology over the past few decades has substantially advanced our knowledge of the pathophysiology of atherosclerosis and athero-thrombosis. The role of the endothelium in maintaining vascular health and the link between endothelial dysfunction and atherosclerosis has been verified. The systemic nature of endothelial dysfunction leading to localized manifestations of atherosclerosis, exacerbated by inflammation is now appreciated. The clinical manifestations of acute coronary syndromes and chronic ischemic coronary artery disease are ultimately a consequence of these processes. The evolution of the acute coronary syndromes is particularly related to the development of intravascular athero-thrombotic disease involving the epicardial coronary vasculature. The recognition of these thrombogenic processes and their sequelae is essential to the management of these syndromes.
Endothelium, endothelial dysfunction and atherosclerosis
The mono-cellular layer of endothelial cells which lines the vascular lumen is critical in maintaining normal vascular flow. The endothelium modulates vascular flow by controlling vasodilator tone and it inhibits platelet aggregation and clotting factor activation by providing a barrier to the procoagulant sub-endothelial tissue. A healthy endothelium also acts as a barrier to inflammation and is able to adequately repair itself after injury. Endothelial dysfunction is manifested by vasoconstriction, thrombosis, inflammation and smooth muscle proliferation. Atherosclerotic lesions seem to develop under an intact but leaky and dysfunctional endothelium. Many traditional coronary risk factors that predispose to atherosclerosis such as hypercholesterolemia, hypertension and a positive family history are associated with endothelial dysfunction.1,2,3 Prospective cohort studies have shown endothelial dysfunction to independently predict progression of atherosclerosis and acute cardiovascular events in patients with and without known coronary artery disease.4,5
Ludmer et al. provided the first evidence of endothelial dysfunction in humans6. They showed paradoxical constriction of the coronary arteries to acetylcholine in individuals with mild as well as severe coronary artery disease, suggesting that endothelial dysfunction is present early in the development of atherosclerosis. Endothelial dysfunction is thought to result from damaging environmental exposure possibly exacerbated by genetic predisposition. Oxidative injury from various metabolic derangements in the local environment, physical forces (shear stress) and even infectious processes may all contribute to endothelial injury. Endothelial dysfunction results in a leaky endothelial lining which allows for passage of pro-atherogenic stimuli into the sub-endothelial space. Later, endothelial cells may vanish and denuded areas appear with exposure of blood to sub-endothelial tissue7. Loss of endothelial function leads to reduced local nitric oxide availability along with increased expression of prothrombotic factors, chemokines and proinflammatory mediators and reduced number and function of endothelial progenitor cells.8 With lower nitric oxide availability there is increased expression of intercellular adhesion molecules by the endothelial cells which leads to the binding of monocytes and lymphocytes, with subsequent invasion of the vascular wall9. The complex local interactions surrounding the damaged endothelium facilitates further passage of lipids and leucocytes into the sub-endothelium. Monocytes transmigrate into the subendothelial space, ingest the oxidized lipoproteins and then are transformed into macrophages. Macrophages accumulate oxidized LDL transforming into foam cells. These foam cells form the initial lesions leading to advanced atherosclerosis10,11 (Figure 1).
Stable and “vulnerable” plaques
Foam cells secrete pro-inflammatory cytokines including growth factors, matrix metalloproteinases and tissue factor leading to migration and proliferation of smooth muscle cells in the lesions10. Recurrent bouts of inflammation induce further smooth muscle cell proliferation and migration into the intima. This can lead to the transformation into a complex lesion consisting of inflammatory cells, smooth muscle cells, and intracellular and extracellular lipid.11, 12 Neovascularization of these lesions with immature vasculature may lead to hemorrhage within the lesions further increasing plaque size and lipid content.13,14,15,16 Generally, a thick layer of smooth muscle cells, collagen and elastin forms a cap that covers the lesion and keeps its contents sequestered from the blood stream. These voluminous lesions cause varying degrees of remodeling of the vessel wall along with varying degrees of stenosis of the intraluminal area. Inflammation leads to worsening atherosclerosis and thinning of the fibrous cap with the potential for subsequent plaque destabilization (Figure 2).
The balance between cell migration, cell proliferation, extracellular matrix formation, inflammatory leucocyte interaction, and cell apoptosis plays a role in the transition from a stable plaque to a potentially high risk or so called “vulnerable” plaque. As to which plaque is likely to become the site of an intraluminal coronary thrombus and become symptomatic has been intensively studied and debated.17 Digestion of the fibrous matrix by matrix metallo-proteinases and apoptosis of the smooth muscle cells forming the caps may lead to weakening and thinning of the fibrous cap. The death of macrophages by apoptosis and necrosis along with plaque hemorrhage from leaky vasa-vasorum contributes to the formation of a soft and destabilizing lipid-rich core within the plaque. Macrophages and T-cells congregating at sites of fibrous cap disruption indicate their key role in plaque destabilization releasing necrotic material into the vessel lumen along with cholesterol crystals. The necrotic sub-endothelial material along with lipid crystals interacts with platelets in the blood leading to coronary thrombus formation. In addition, tissue factor that is exposed with plaque disruption results in activation of the extrinsic coagulation pathway.
Following plaque disruption, the plaque contents come in contact with blood. Freely circulating platelets in circulation promptly adhere to the sub-endothelial matrix and damaged endothelial cells. Adhesion of platelets is mediated by the binding of surface glycoproteins to endothelial ligands. Platelet surface glycoprotein GP Ib/IX recognizes von-Willebrand factor synthesized and stored in endothelial cells. Platelet glycoprotein GPIa/IIa binds collagen present in the deeper vessel wall. Following adhesion, activated platelets release multiple intermediaries including serotonin, ADP, thromboxane A2, endothelin, free radicals and platelet activating factor which promote further platelet aggregation and vasoconstriction. These platelet aggregates forming a so called white thrombus tend to be unstable and may cause intermittent reduction in blood flow and distal embolization18. The coagulation status of blood is a significant determinant of the end result of plaque disruption. Rapid activation of the coagulation system with fibrin deposition strengthens the platelet aggregates causing persistent obstruction to flow. This along with impaired flow dynamics from vasoconstriction leads to pooling of blood and formation of fibrin rich red thrombi.19
Clinical manifestations of athero-thrombotic processes in coronary arteries
The atherosclerotic processes involving the vasculature evolve over many years (Figure 3). Early plaque formation is often associated with outward remodeling of the vessel. When this adaptive process is exceeded, the plaque starts to encroach into the lumen of the vasculature. It is now recognized that thrombosis from plaque disruption or erosion of a large plaque that is not severely occlusive prior to the event is most often the immediate cause of acute coronary syndromes, particularly ST elevation myocardial infarction.20 Plaque disruption is found at pathology in 10% of individuals with atherosclerosis dying of non-cardiac disease. Conversely, thrombi are frequently observed at sites other than those of the major culprit lesion in patients dying of acute coronary syndromes21. Thus, thrombus formation on a plaque may or may not lead to a clinical syndrome. While plaque disruption with thrombus formation is thought to be the major pathogenetic mechanism for acute coronary syndromes, the vast majority of plaque fissures are asymptomatic and may only contribute to the slow progression of atherosclerotic lesions22. While plaques associated with acute myocardial infarction are usually large, expansively remodeled and not severely narrowed prior to the clinical event, plaques responsible for stable angina usually are smaller but, often are associated with more severe luminal narrowing because of concomitant constrictive remodeling.23-25
About 70% of cases of acute coronary thrombosis involve a disrupted atherosclerotic plaque and in the remaining 30%, there is only superficial intimal injury at the site of thrombus formation.26 Superficial endothelial erosion leading to a clinically evident coronary thrombosis is most commonly seen in women and in diabetics with hypertriglyceridemia. While the exact mechanism of superficial erosion is not clear, it is likely that matrix metallo-proteinases in the subendothelium may disrupt the tethering of the endothelial cell to the basal lamina leading to desquamation.27 Many cases are likely associated with a prothrombotic milieu. Most episodes of endothelial erosion are also likely asymptomatic. However erosion may lead to non-occlusive thrombus formation followed by healing. Such repetitive cycles could contribute to slow atherosclerotic progression.
Determinants of the clinical syndrome
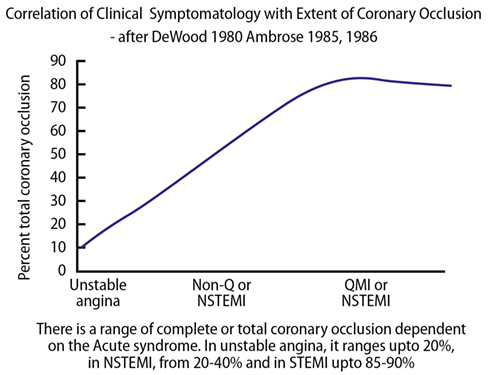
Apart from the characteristics and volume of the plaque content and its location in the coronary tree, it is, 1- the cellular and humoral components of blood, 2- the extent and type of thrombus formation at the site of plaque disruption/erosion and 3- myocardial vulnerability in the individual that contributes to the extent and type of clinical manifestation if any. The syndrome ultimately developed also depends on factors such as the degree and acuteness of obstruction, the duration of decreased perfusion and the relative myocardial oxygen demand as well as the collateral circulation28. Angiographic, biochemical, pharmacologic and surgical data support the role of thrombus formation in patients presenting with STEMI, NSTEMI and unstable angina.20,29 When unstable angina is defined as the new onset of low work load or rest angina or an abrupt change in angina that had previously been stable, the incidence of finding a complex plaque (i.e. an eccentric stenosis with overlapping edges, irregular borders, ulcerations and/or filling defects in the culprit vessel) is approximately 70% on angiographic analysis30. In patients with a short duration of unstable angina or with very recent onset of rest pain, the incidence of thrombus has been reported to be even higher.31 Coronary angioscopy has demonstrated an even higher incidence of mural thrombus in patients presenting with unstable angina.32,33 In unstable angina and non-Q wave myocardial infarction (the forerunner of NSTEMI), the thrombus is more likely to be non-occlusive than in an evolving Q-wave myocardial infarction or STEMI, where the thrombus in the first few hours after infarction is occlusive in greater than 80%29 (Figure 4). Not all patients with unstable angina/NSTEMI necessarily have plaque disruption/erosion. Type II myocardial infarction related to a supply/demand mismatch34 accounted for about 30% of all myocardial infarction in a prospective study by Javed et al.35
Coronary thrombus and PCI
Pathological findings of thrombus collected by thrombectomy procedures during PCI in the last few years corroborate the role of in situ thrombus in acute coronary syndromes. Histopathological analysis of aspirated thrombotic content show erythrocyte-rich (red) thrombus in about 35% of patients, predominantly in those presenting with low TIMI flow. A platelet-rich thrombus is identified in 65% of cases, particularly in the early hours of acute myocardial infarction36. It has also come to light that the composition of thrombus is often heterogenous. The composition of the isolated thrombus often shows fresh thrombus along with features of organization, and lytic changes in the same tissue fragment37. The layered composition of clots suggests episodic growth of thrombus for a finite interval before the onset of occlusive thrombus and clinical symptomatology. Analysis of electron microscopic images of thrombus obtained from thrombectomy procedures shows that formation of the thrombus is dynamic and that the composition of the thrombus varies with the ischemia time. Fresh thrombi have a highest proportion of platelets, whereas the proportion of fibrin fibers increases over time leading to older more fibrin rich thrombi38 (Figure 5).
The thrombus burden associated with an acute coronary syndrome may vary depending on various factors, including size of the vessels, duration of occlusion, prothrombotic state etc. Some of the clinical risk factors associated with higher thrombus burden include hypercholesterolemia, smoking and male gender.39 In addition, a variety of local factors affect the genesis and behavior of thrombus generated in situ including interaction between platelets, vessel wall, red blood cells, plaque gruel and coagulation proteins. The behavior of thrombus during percutaneous intervention may be quite variable and may influence the outcome of the intervention. The extent and duration of fibrin polymerization and stabilization in an evolving thrombus may contribute to the differing behavior of thrombus including the tendency for friability during catheter or wire maneuvering despite rigid adherence to the underlying plaque.
The fibrin network in thrombi, when examined by electron microscopy shows two distinct types of patterns.40 One pattern consists of dense scaffolding of thin fibers that is resistant to mechanical force and thrombolysis as compared to the second pattern consisting of thick loosely packed fibers that are more susceptible to thrombolysis. Interactions between platelets, red blood cells, vessel wall, fibrinogen and other local chemicals in the local environment in addition to the age of the thrombus may all have an impact on the fibrin network and thus on the strength and behavior of a thrombus. The characteristics of the clots as seen on angiography correlate with the histology of extracted thrombus.41,42 The slightly altered behavior of coronary thrombus in the setting of tobacco use might be explained by differences in the fibrin architecture as seen by electron microscopy.43
Other factors that can influence clot burden and behavior include characteristics of the underlying vessel; notably coronary arteries with ectasia, vasculitis and aneurysms are more likely to have large thrombi due to stasis as do saphenous vein grafts. Late presentation with an established myocardial infarction and cardiogenic shock, inadequate anti-coagulant or anti-platelet therapy and complications related to therapy such as HIT may also increase thrombus burden. The right coronary artery tends to have a larger burden of thrombus probably because of proximal propagation of thrombus related to fewer branch points. Hyperglycemia and leukocytosis may also accentuate thrombosis. During PCI, thrombus growth may be triggered by guidewires, stasis of blood, inadequate antithrombotic/anticoagulant therapy, balloons or stent; the “angry clot” phenomenon.44, 45
Complications of PCI Related to Intracoronary Thrombus
Prior to the introduction of stents and methods to extract or dissolve thrombus, balloon angioplasty of thrombotic lesions posed many procedural difficulties for the interventionalist including major dissection, vasoconstriction, abrupt closure and total occlusion with the need for emergent bypass and was associated with an increased mortality.46,47 Pre-existing thrombus was also shown to increase angiographic restenosis, mainly through early vessel occlusion.48 Pre-existing thrombus continued to be an independent predictor of angioplasty failure until the introduction of stents. While many of these procedural complications have been significantly reduced in the stent era, there continue to be adverse complications of intracoronary thrombus during PCI and stenting related to embolization, the “no reflow” phenomenon and stent thrombus. The limited success of standard PCI even with stents, in lesions with a large thrombus burden despite the illusion of adequate reperfusion is clinically relevant and contributes to the no-reflow phenomenon.49 Of note, in some studies the desired grade 3 myocardial blush score was gained only in a minority of patients in whom TIMI 3 flow was established with standard PCI at the time of primary PCI.50
The first large scale study of the effect of thrombus burden during PCI was reported by Harjai et al.51 About 6% of patients in the study that included 2,148 subjects from PAMI-2, Stent PAMI and PAMI No-Surgery-On-Site trials had angiographically visible thrombus after PCI. The presence of thrombus before PCI was independently associated with a higher incidence of post-PCI thrombus likely reflecting a larger thrombus burden prior to intervention. Both the thrombus burden and thrombus characteristics play a role in contributing to the no-reflow phenomenon. The higher thrombus burden can lead to more distal embolization obstructing flow within distal arterial segments and microvessels leading to no-reflow.52 Yip et al. proposed a score to assess thrombus burden on the basis of angiographic features (Table 3).53
All of these features were independent predictors of no-reflow in 800 patients undergoing primary PCI. Using Yip’s score, Limbruno et al were able to predict total debris volume captured by distal filter wire in patients undergoing PCI for STEMI.54 Patients with no-reflow following PCI for STEMI have also been shown to have decreased clot permeability with increased resistance to lysis of thrombus when compared to patients with more normal flow.55 Of note, distal embolization of thrombotic debris often occurs after stent deployment in large coronary vessels, whereas in small vessels it is possible that the stent itself might fix the thrombus to the vessel wall.
Patients who develop no-reflow related to distal embolization sustain larger infarcts leading to poorer outcomes56. Distal embolization occurs in upto 15% of patients undergoing PCI and is associated with as much as a seven fold increase in the rate of periprocedural MI. In more recent trials with concurrent use of anticoagulants and dual antiplatelet therapy the incidence of distal embolization is approximately 6%.57 These complications can be seen more often in patient undergoing PCI for acute myocardial infarction.58 Individuals with angiographically evident embolization have significantly worse outcomes than patients without, as expressed by lower myocardial blush grade (MBG), impaired ST-segment resolution, and higher level of myocardial enzyme leakage.59-64 Thrombus embolization also significantly increases the need for emergency bypass surgery as well as the procedure-related death rate.65
No-reflow in the setting of a large thrombus can be assessed with TIMI flow grade and MBG during PCI. Clinically it can be suspected by lack of ST segment resolution. Noninvasive imaging techniques such as myocardial contrast echocardiography and cardiac magnetic resonance imaging provide a more accurate assessment of myocardial perfusion and hence a better assessment of no-reflow following PCI.66,67 Direct stenting has been suggested as a technique to reduce distal embolization, by avoiding balloon-induced thrombus fragmentation and entrapment of thrombus under the stent struts.68 However, direct stenting is feasible only in patients with good distal visualization of infarct related artery after passage of a guidewire.
Thrombectomy devices and distal filters were developed as approaches to prevent no-reflow from thrombus embolization. The negative results in two large trials, one involving rheolytic thrombectomy and another involving distal occlusive protection, have tempered the adoption of these technologies.69, 70 The negative results in these trials may have been because of poor selection of patients; that is these techniques were used indiscriminately in all comers including patients at low risk of no-reflow.
Aspiration thrombectomy was developed as a technique for thrombus extraction following the early disappointing results of rheolytic thrombectomy. A more recent report on manual thrombus-aspiration by Svilaas et al (TAPAS) was a landmark study. The TAPAS investigators, studied the sequelae of angiographically visible distal embolization after PCI in patients presenting with STEMI.71 This open labeled study involved 1071 patients undergoing PCI who were randomized to aspiration thrombectomy versus conventional PCI. Thrombus aspiration resulted in improved myocardial perfusion, indicated by the myocardial blush grade and ST-segment analysis on the 12-lead electrocardiogram, compared with conventional PCI. A follow up report on this cohort found that a strategy of thrombus aspiration before stenting during primary PCI resulted in a lower cardiac mortality and a lower incidence of the combined endpoint of cardiac death or non-fatal reinfarction than normal therapy alone58 Thus, of the various thrombectomy procedures developed thus far, manual thrombus aspiration has shown a favorable clinical outcome with reduction in incidence of both distal emboli and mortality.72 Based in the results of the above and other such trials, thrombus extraction devices have been approved for use in the most recent focused update of guidelines on management of patients with STEMI.73
A recent study by Migliorini et al. reported on the use of rheolytic thrombectomy with Angiojet. In this multicenter, prospective, RCT, 501 patients with acute myocardial infarction, angiographic evidence of thrombus grade 3 to 5, and a reference vessel diameter > 2.5 mm were randomized to direct stenting alone or direct stenting along with thrombectomy. There was no difference between the two treatment strategies with respect to the coprimary end points of ST-segment elevation resolution and scintigraphic infarct size. Nevertheless in terms of clinical outcomes, significant differences were observed in favor of the mechanical thrmobectomy arm regarding the composite of major adverse events at 6 and 12 months adding new arguments in support of rheolytic thrombectomy.74
It will be relevant to re-emphasize that the benefit of any thrombectomy procedure during primary PCI is likely dependent on the thrombus burden as supported by the findings of the REMEDIA trial. The REMEDIA (Randomized Evaluation of the Effect of Mechanical Reduction of Distal Embolization by Thrombus-Aspiration in Primary and Rescue Angioplasty) trial, which was the first randomized trial to assess the role of thrombectomy performed with a simple manual aspiration catheter, as compared with conventional PCI showed better myocardial perfusion indices as compared with standard primary PCI.75 This benefit was particularly noted in subjects with higher thrombus burden emphasizing the importance of proper patient selection in the use of these techniques.
Stent thrombosis
The etiology of stent thrombosis is multifactorial, and includes stent thrombogenicity, in addition to procedure related, lesion related, and patient-related factors.76 Randomized trials of BMS implantation during elective PCI have reported stent thrombosis rate of 0.4 to 1.3%.77,78 Results from a pooled analysis of 10 randomized controlled trials of elective PCI reported similar rates of stent thrombosis between BMS and DES of around 0.6%; but most studies indicate a higher rate of very late stent thrombosis with DES compared to BMS, particularly for the first generation DES.79-82 Stent underexpansion, malapposition, residual dissections, and inflow/outflow disease have been well established by intravascular ultrasound as mechanical causes related to early stent thrombosis for both BMS and DES.83-86 However, by and far premature discontinuation of dual anti-platelet therapy remains the most frequent but not the only cause of stent thrombosis.87
Apart from mechanical factors related to the stents, there are other variables including alterations in the coagulation cascade and response to pharmacotherapy that may influence the behavior of thrombus during PCI, particularly in the setting of STEMI. Acute coronary syndromes by their very evolution have intracoronary thrombus and have been associated with higher rates of stent thrombosis following PCI. In the TYPHOON (Trial to Assess the Use of the Cypher Stent in Acute Myocardial Infarction Treated With Angioplasty) trial, which randomized STEMI patients to SES or BMS, the angiographic stent thrombosis rate was 2.0% and 3.4%, respectively, at 1 year.88 Clearly inadequate anticoagulation and antiplatelet therapy during PCI is associated with increased thrombus burden during PCI and consequent stent thrombosis. An impaired response to antiplatelet therapy may also predispose to a large thrombus burden and consequent stent thrombus, particularly in the setting of ACS.89,90 The impaired response may be in the form of resistance to either aspirin, clopidogrel or side effects of medications such as in the case of heparin- induced thrombocytopenia.
Along with other causes, the presence of thrombus has been identified as a factor predisposing to stent thrombosis.76 Thrombus that is compressed against the vessel wall and or displaced by stent struts during primary PCI may cause problems in the long term due to resolution of thrombus leaving behind a malapposed stent. Thus a larger thrombotic burden during PCI may increase the risk of sent thrombosis due to late malapposition. This was shown by Sianos et al in a retrospective cohort of 812 consecutive patients presenting with STEMI who had primary PCI with use of a drug eluting stent.91 A large thrombus burden was a fundamental factor for adverse clinical outcomes including increased 30 day mortality, with high rates of infarct-related stent thrombosis accounting for majority of the post-30-days MACE rate. Large thrombus burden in that study was defined as a filling defect whose length was greater than or equal to twice the vessel diameter. In patients with total occlusion of the infarct artery, reperfusion was established with a wire or small balloon before thrombus burden was assessed; this was an important aspect of the methodology since a simple classification based on TIMI flow would have excluded a majority of the subjects who would have presented with TIMI flow of zero. The authors found that the risk of subsequent stent thrombosis after primary PCI with stenting can be dramatically reduced with rheolytic thrombectomy.
In summary intra-coronary thrombogenesis is a dynamic process and the thrombus burden in the coronary vasculature during an acute coronary event is variable. The factors that influence the burden and behavior of the thrombus are complex. Elaboration of these processes has led to the recognition and improved anticipation of their consequences.
Conclusion
Endothelial dysfunction and atherosclerosis set the stage for coronary artery disease. Acute clinical manifestations occur from destabilization of atherosclerotic plaques within the coronary vessels, most often secondary to intraluminal thrombus formation. Appreciating the role of thrombus formation in ACS has lead to a significant evolution in the management of these challenging patients. Thrombus during percutaneous intervention poses a formidable challenge for the interventionalist both in terms of dealing with the possibility of embolization and no-reflow in the acute situation and stent thrombus acutely and in the long term. Though thrombus may be detected by various imaging modalities including angiography, ultrasound, and OCT, angiographic techniques have been standardized and still remain the initial modality that is routinely used for decision making during PCI especially in the setting of STEMI and other NSTEACS. Several techniques both pharmacologic and mechanical have been developed to deal with intra-coronary thrombus. A general understanding of the processes involved in athero-thrombosis and its treatment is essential in maximizing patient outcomes.
-
John S, Schlaich M, Langenfield M, et al. Increased bioavailability of nitric oxide after lipid-lowering therapy in hypercholesterolemic patients: a randomized, placebo-controlled, double-blind study. Circulation 1998; 98:211-216.
-
Vita JA, Treasure CB, Nabel EG, et al. Coronary vasomotor response to acetylcholine relates to risk factors for coronary artery disease. Circulation 1990;81:491-497.
-
Schächinger V, Britten MB, Elsner M, et al. A positive family history of premature coronary artery disease is associated with impaired endothelium-dependent coronary blood flow regulation. Circulation 1999;100: 1502-1508.
-
Schächinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction and adverse long-term outcome of coronary heart disease. Circulation 2000; 101: 1899-1906.
-
Halcox JPJ, Schenk WH, Zalos G, et al. Prognostic value of coronary vascular endothelial function. Circulation 2002; 106: 653-658].
-
Ludmer PL, Selwyn AP, Shook TL, et al. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315:1046-1051.
-
Davies MJ, Woolf N, Rowles PM, Pepper J. Morphology of the endothelium over atherosclerotic plaques in human coronary arteries. Br Heart J 1988;60:459–64.
-
Quyyumi AA. Prognostic value of endothelial function. Am J Cardiol 2003; 91:19H-24H.
-
Khan BV, Harrison DG, Olbrych MT, et al. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc Natl Acad Sci USA 1996; 93(17):9114-9119.
-
Libby P. Inflammation in atherosclerosis. Nature 2002; 420: 868 -874.
-
Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 2005;352:1685–95.
-
Schwartz SM, Virmani R, Rosenfeld ME. The good smooth muscle cells in atherosclerosis. Curr Atheroscler Rep 2000;2:422–9.
-
Barger AC, Beeuwkes R 3rd, Lainey LL, Silverman KJ. Hypothesis: vasa vasorum and neovascularization of human coronary arteries. A possible role in the pathophysiology of atherosclerosis. N Engl J Med 1984;310:175–177.
-
Kolodgie FD, Gold HK, Burke AP, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med 2003;349:2316 –25.
-
Virmani R, Kolodgie FD, Burke AP, et al. Atherosclerotic plaque progression and vulnerability to rupture angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol 2005;25:2054–61.
-
Casscells W, Hassan K, Vaseghi MF, et al. Plaque blush, branch location, and calcification are angiographic predictors of progression of mild to moderate coronary stenoses. Am Heart J 2003;145:813–20.
-
Ambrose JA. In search of the “vulnerable plaque”. Can it be localized and will focal regional therapy ever be an option for cardiac prevention? J Am Coll Cardiol. 2008;51:1539-1542.
-
Maseri A, Chierchia S, Davies G. Pathophysiology of coronary occlusion in acute infarction. Circulation 1986; 68:28B-35B.
-
Schwartz SM, Galis SG, Rosenfeld ME, et al. Plaque rupture in humans and mice. Arterioscler Thromb Vasc Biol. 2007;27:705-713.
-
Ambrose JA, Tannenbaum MA, Alexopoulos D, et al. Angiographic progression of coronary artery disease and the development of myocardial infarction. J Am Coll Cardiol 1988; 12:56-62.
-
Davies MJ. The birth, growth and consequences of the atherosclerotic plaque. Dialogues Cardiovasc Med 1999; 4:115-178.
-
Ip JH, Fuster V, Badimon L, Badimon J, Taubman MB, Chesebro JH. Syndromes of accelerated atherosclerosis: role of vascular injury and smooth muscle proliferation. J Am Coll Cardiol 1990; 15: 1667-87.
-
Vink A, Schoneveld AH, Richard W, et al. Plaque burden, arterial remodeling and plaque vulnerability: determined by systemic factors? J Am Coll Cardiol 2001;38:718 –23.
-
Ambrose JA. Plaque disruption and the acute coronary syndromes of unstable angina and myocardial infarction: if the substrate is similar, why is the clinical presentation different? J am Coll Cardiol 1992; 19(7): 1653-8.
-
Ambrose JA, Weinrauch M. Thrombosis in ischemic heart disease. Arch Intern Med 1996; 156(13): 1382-94.
-
Farb A, Burke AP, Tang A, et al. Coronary plaque erosion without rupture into a lipid core: a frequent cause of coronary thrombosis in sudden coronary death. Circulation. 1996;93:1354-1363.
-
Sugiyama S, Kugiyama K, Aikawa M, et al. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: Involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler Thromb Vasc Biol 2004; 24: 1309-14.
-
Gorlin R, Fuster V, Ambrose JA. Anatomic-physiologic links between acute coronary syndromes. Circulation 1986; 74: 6-9.
-
DeWood MA, Spores J, Notske R, et al. Prevalence of total coronary occlusion during the early hours of transmural myocardial infarction. N Engl J Med 1980; 303: 897-902.
-
Ambrose JA, Winters SL, Arora RR, et al. Angiography evolution of coronary artery morphology in unstable angina. J Am Coll Cardiol 1986;7(3):472-478.
-
Capone G. Wolf NM. Meyer H. Meister SG. Frequency of intracoronary filling defects by angiography in angina pecroris at rest. Am J Cardiol 1985; 56:403-6.
-
Sherman CT, Litvack F. Grundfest W, et al. Coronary angioscopy in patients with unstable angina pectoris. N Engl J Med 1986;315: 913-9.
-
Ramee SR, White CJ, Collins TJ, Mesa JE, Murgo JP. Percutaneous angioscopy during coronary angioplasty using a steerable microangioscope. J Am Coll Cardiol 1991; 17:100-5.
-
Thygesen K, Alpert JS, White HD; Joint ESC/ACCF/AHA/WHF Task Force for the redefinition of myocardial infarction, et al. Universal definition of myocardial infarction. Circulation 2007; 116(22): 2634-53.
-
Javed U, Aftab W, Ambrose JA, et al. Frequency of elevated Troponin I and diagnosis of acute myocardial infarction. Am J Cardiol 2009; 104(1): 9-13.
-
Vlaar PJ, Svilaas T, Vogelzang M, et al. A Comparison of 2 thrombus aspiration devices with histopathological analysis of retrieved material in patients presenting with ST-segment elevation myocardial infarction. JACC Cardiovasc Intervent. 2008; 1:265-267.
-
Rittersma S, et al. Plaque instability frequently occurs days or weeks before occlusive coronary thrombosis. Circulation 2005;111:1160-1165.
-
Silvian J, et al. Composition of coronary thrombus in acute myocardial infarction. J Am Coll Cardiol 2011; 57: 1359-67.
-
Zhao XQ, Theroux P, Snapinn SM, Sax FL. Intracoronary thrombus and platelet glycoprotein IIb/IIIa receptor blockade with tirofiban in unstable angina or non-Q-wave myocardial infarction: angiographic results from the PRISM- PLUS trial (Platelet Receptor Inhibition for Ischemic Syndrome Management in Patients Limited by Unstable Signs and Symptoms) Circulation 1999;100: 1609-15.
-
Carr ME, Carr SL. Fibrin structure and concentration alter clot elastic modulus but do not alter platelet medicated force development. Blood Coag Fibrinolysis. 1995;6:79-86.
-
Beygui F, Collet JP, Nagaswami C, et al. Architecture of intracoronary thrombi in ST-elevation acute myocardial infarction: time makes the difference. Circulation. 2006;113:e21-e23.
-
GS, Eisenberg JD, Mittleman MA, et al. Detecting and differentiating white from red coronary thrombus by angiography in angina pectoris and in acute myocardial infarction. Am J Cardiol. 1999;83:94-97.
-
Barua RS, Sy F, Srikanth S, et al. Effects of cigarette smoke exposure on clot dynamics and fibrin structure: an ex vivo investigation. Arterioscler Thromb Vasc Biol. 2010; 30:75-79.
-
Topaz O. On the hostile massive thrombus and the means to eradicate it. Catheterization and Cardiovascular Interventions. 2005; 65(2): 280-281.
-
Saber RS, Edwards WD, Bailey KR, McGovern TW, Schwartz RS, Holmes D. Coronary embolization after balloon angioplasty or thrombolytic therapy: an autopsy study of 32 cases. J Am Coll Cardiol 1993;22:1283–1288.
-
White CJ, Ramee SR, Collins TJ, et al. Coronary thrombi increase PTCA risk: angioscopy as a clinical tool. Circulation 1996;93: 253–258.
-
Singh M, Berger PB, Ting HH, et al. Influence of coronary thrombus on outcome of percutaneous coronary angioplasty in the current era (the Mayo Clinic Experience). Am J Cardiol 2001;88:1091–1096.
-
Violaris AG, Melkert R, Herrman JP, Serruys PW. Role of angiographically identifiable thrombus on long-term luminal renarrowing after coronary angioplasty: a quantitative angiographic analysis. Circulation 1996;93:889–897.
-
Rezkalla SH, Kloner RA. Coronary no-reflow phenomenon: from the experimental laboratory to the cardiac catheterization laboratory. Catheter Cardiovasc Interv 2008;72:950–957.
-
Kaya MG, Arsian F, Abaci A, et al. Myocardial blush grade: a predictor for major adverse cardiac events after primary PTCA with stent implantation for acute myocardial infarction. Acta Cardiol. 2007;62:445-451.
-
Harjai KJ, Grines C, Stone GW, Boura J, Turco M, Brodie B, Sadeghi M, Cox D, Grines L, O’ Neill, WW on behalf of the Primary Angioplasty in Myocardial Infarction (PAMI) Investigators. Am J Cardiol 2003; 92:377-382.
-
Henriques JPS, Zijlstra F, Ottervanger JP, de Boer MJ, et al. Incidence and clinical significance of distal embolization during primary angioplasty for acute myocardial infarction. Eur Heart J 2002; 23:1112-1117.
-
Yip HK, Chen MC, Chang HW, et al. Angiographic morphologic features of infarct-related arteries and timely reperfusion in acute myocardial infarction: predictors of slow-flow and no-reflow phenomenon. Chest 2002;122:1322–32.
-
Limbruno U, De Carlo M, Pistolesi S, et al. Distal embolization during primary angioplasty: histopathologic features and predictability. Am Heart J 2005;150:102– 8.
-
Zalewski J, Undas A, Godlewski J, Stepien E, Zmudka K. No-reflow phenomenon after acute myocardial infarction is associated with reduced clot permeability and susceptibility to lysis.
-
G, Kleinbongard P, Bose D, et al. Coronary microembolization: from bedside to bench and back to bedside. Circulation. 2009;120(18):1822-1836.
-
Fokkema ML, Vlaar PJ, Svilaas T, Vogelzang M, et al. Incidence and clinical consequences of distal embolization on the coronary angiogram after percutaneous coronary intervention for ST-elevation myocardial infarction. Eur Heart J 2009; 30: 908-915.
-
Vlaar PJ, Svilaas T, van der Horst IC, et al. Cardiac death and reinfarction after 1 year in the Thrombus Aspiration during Percutaneous coronary intervention in Acute Myocardial infarction Study (TAPAS): a 1-year follow-up study. Lancet 2008; 371:1915-20.
-
Brosh D, Assali AR, Mager A, et al. Effect of no-reflow during primary percutaneous coronary intervention for acute myocardial infarction on six-month mortality. Am J Cardiol 2007;99:442–5.
-
Henriques JP, Zijlstra F, van’t Hof AW, et al. Angiographic assessment of reperfusion in acute myocardial infarction by myocardial blush grade. Circulation 2003;107:2115–9.
-
Gibson CM, Cannon CP, Murphy SA, Marble SJ, Barron HV, Braunwald E, TIMI Study Group. Relationship of the TIMI myocardial perfusion grades, flow grades, frame count, and percutaneous coronary intervention to long-term outcomes after thrombolytic administration in acute myocardial infarction. Circulation 2002; 105:1909 –13.
-
McLaughlin MG, Stone GW, Aymong E, et al. Prognostic utility of comparative methods for assessment of ST-segment resolution after primary angioplasty for acute myocardial infarction: the Controlled Abciximab and Device Investigation to Lower Late Angioplasty Complications (CADILLAC) trial. J Am Coll Cardiol 2004;44: 1215–23.
-
Bolognese L, Carrabba N, Parodi G, et al. Impact of microvascular dysfunction on left ventricular remodeling and long-term clinical outcome after primary coronary angioplasty for acute myocardial infarction. Circulation 2004,109:1121– 6.
-
Galiuto L, Garramone B, Scarà A, et al., AMICI Investigators. The extent of microvascular damage during myocardial contrast echocardiography is superior to other known indexes of post-infarct reperfusion in predicting left ventricular remodeling: results of the multicenter AMICI study. J Am Coll Cardiol 2008;51:552–9.
-
T, Ueda Y, Shimizu M, et al. Association between cardiac troponin T elevation and angioscopic morphology of culprit lesion in patients with non-ST segment elevation acute coronary syndrome. Am Heart J. 2005;150:227-233.
-
Galiuto L, Garramone B, Scarà A, et al., AMICI Investigators. The extent of microvascular damage during myocardial contrast echocardiography is superior to other known indexes of post-infarct reperfusion in predicting left ventricular remodeling: results of the multicenter AMICI study. J Am Coll Cardiol 2008;51:552–9.
-
Wu KC, Zerhouni EA, Judd RM, et al. Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation 1998;97:765–72.
-
Loubeyre C, Morice MC, Lefèvre T, Piéchaud JF, Louvard Y, Dumas P. A randomized comparison of direct stenting with conventional stent implantation in selected patients with acute myocardial infarction. J Am Coll Cardiol 2002;39:15–21.
-
Ali A, Cox D, Dib N, et al., AIMI Investigators. Rheolytic thrombectomy with percutaenous coronary intervention for infarct size reduction in acute myocardial infarction: 30-day results from a multicenter randomized study. J Am Coll Cardiol 2006; 48:244-52.
-
Dangas G, Stone GW, Weingerg MD, et al., EMERALD Investigators. Contemporary outcomes of rescue percutaneous coronary intervention for acute myocardial infarction: comparison with primary angioplasty and the role of distal protection devices (EMERALD trial). Am Heart J 2008; 155: 1090-6.
-
Svilaas T, Vlaar PJ, van de Horst IC, et al. Thrombus aspiration during percutaneous coronary intervention in acute myocardial infarction. N Engl J Med 2008; 358:557-67.
-
De Luca G, Dudek D, Sardella G, Marino P, Chevalier B, Zijlstra F. Adjunctive manual thrombectomy improves myocardial perfusion and mortality in patients undergoing primary percutaneous coronary intervention for ST-elevation myocardial infarction: a meta-analysis of randomized trials. Eur Heart J 2008;29:3002-10.
-
Kushner FG, Hand M, Smith SC Jr., et al. 2009 focused updates: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update). A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2009; 54:2205-41.
-
Migliorini A, Stabile A, Rodriquez AE, et al.: JETSTENT Trial Investigators. Comparison of Angiojet rheolytic thrombectomy before direct infarct artery stenting with direct stenting alone in patients with acute myocardial infarction. The JETSTENT trial. J Am Coll Cardiol 2010;56(16): 1298-306.
-
Burzotta F, Trani C, Romagnoli E, et al. Manual thrombus aspiration improves myocardial reperfusion: the randomized evaluation of the effect of mechanical reduction of distal embolization by thrombus-aspiration in primary and rescue angioplasty (REMEDIA) trial. J Am Coll Cardiol 2005; 46:371-6.
-
Cheneau E, Leborgne L, Mintz GS, et al. Predictors of subacute stent thrombosis. Results of a systematic intravascular ultrasound study. Circulation 2003; 108:43–47.
-
Leon MB, Baim DS, Popma JJ, et al. A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting. Stent Anticoagulation Restenosis Study Investigators. N Engl J Med 1998; 339:1665–71.
-
Mak KH, Belli G, Ellis SG, Moliterno DJ. Subacute stent thrombosis: evolving issues and current concepts. J Am Coll Cardiol 1996;27:494–503.
-
Moreno P, Fernandez C, Hernandez R, et al. Drug-eluting stent thrombosis: results from a pooled analysis including 10 randomized studies. J Am Coll Cardiol 2005;45:954 –9.
-
Wenaweser P, Daemen J, Zwahlen M, van Domburg R, Jüni P, Vaina S, Hellige G, Tsuchida K, Morger C, Boersma E, Kukreja N, Meier B, Serruys PW, Windecker S. Incidence and correlates of drug-eluting stent thrombosis in routine clinical practice. 4-year results from a large 2-institutional cohort study. J Am Coll Cardiol. 2008;52(14):1134.
-
Airoldi F, Colombo A, Morici N, et al. Incidence and predictors of drug-eluting stent thrombosis during and after discontinuation of thienopyridine treatment. Circulation. 2007;116(7):745.
-
de la Torre-Hernández JM, Alfonso F, Hernández F, et al. Drug-eluting stent thrombosis: results from the multicenter Spanish registry ESTROFA (Estudio ESpañol sobre TROmbosis de stents FArmacoactivos). J Am Coll Cardiol. 2008;51(10):986.
-
Cheneau E, Leborgne L, Mintz GS, et al. Predictors of subacute stent thrombosis: results of a systematic intravascular ultrasound study. Circulation 2003;108:43–7.
-
Cutlip DE, Baim DS, Ho KK, et al. Stent thrombosis in the modern era: a pooled analysis of multicenter coronary stent clinical trials. Circulation 2001;103:1967–71.
-
Uren NG, Schwarzacher SP, Metz JA, et al. Predictors and outcomes of stent thrombosis: an intravascular ultrasound registry. Eur Heart J 2002;23:124 –32.
-
Fujii K, Carlier SG, Mintz GS, et al. Stent underexpansion and residual reference segment stenosis are related to stent thrombosis after sirolimus-eluting stent implantation: an intravascular ultrasound study. J Am Coll Cardiol 2005;45:995– 8.
-
D’Ascenzo F, Bollati M, Clementi F, et al. Incidence and predictors of coronary stent thrombosis: Evidence from an international collaborative meta-analysis including 30 studies, 221,066 patients, and 4276 thromboses. Int J Cardiol 2012 Feb 21 [http://dx.doi.org/10.1016/j.ijcard.2012.01.080].
-
Spaulding C, Henry P, Teiger E, et al. Sirolimus-eluting versus uncoated stents in acute myocardial infarction. N Engl J Med 2006;355:1093–10.
-
Borna C, Lazarowski E, van Heusden C, Ohlin H, Erlinge D. Resistance to aspirin is increased by ST-elevation myocardial infarction and correlates with adenosine diphosphate levels. Thromb J 2005;3:10.
-
Wenaweser P, Dorffler-Melly J, Imboden K, et al. Stent thrombosis is associated with an impaired response to antiplatelet therapy. J Am Coll Cardiol 2005;45:1748 –52.
-
Sianos G, Papafaklis MI, Daemen J, et al. Angiographic stent thrombosis after routine use of drug-eluting stents in ST-segment elevation myocardial infarction: The importance of thrombus burden. J Am Coll Cardiol 2007;50:573-83.
Interventional Cardiology Fellow, UCSF Fresno..
Professor of Medicine, University of California San Francisco. Chief of Cardiology, UCSF Fresno..
Autor correspondencia
Professor of Medicine, University of California San Francisco. Chief of Cardiology, UCSF Fresno..
Correo electrónico: jamambrose@yahoo.com
Para descargar el PDF del artículo
Coronary thrombus: pathophysiology and consequences
![]() Haga click aquí
Haga click aquí
Para descargar el PDF de la revista completa
Revista Argentina de Cardioangiología intervencionista, Volumen Año 2012 Num 01

Revista Argentina de Cardioangiología intervencionista
Número 01 | Volumen
2 | Año 2012

Revista Argentina de Cardioangiolog...
Alfredo Rodríguez
Coronary thrombus: pathophysiology ...
Sundararajan Srikanth y cols.
Bifurcaciones arteriales: ¿cuál t...
Teodoro Bass y cols.
El lado oscuro de la luna: perfil d...
Gastón A Rodríguez-Granillo
Resultados intrahospitalarios en pa...
Carlos Fernández Pereira y cols.
Artículo especial: visión sobre l...
Walter González
Tratamiento de fuga paravalvular mi...
Carlos A Deluca y cols.
Disección espontánea de arteria r...
Martín Bodoira y cols.
Tercera tentativa de recanalizació...
R Pauletto y cols.
Etiquetas
coronary thrombus, percutaneous intervention, endothelial dysfunction, atherosclerosis
Tags
trombo coronario, intervención percutánea, disfunción endotelial, aterosclerosis

Coronary thrombus: pathophysiology and consequences
Autores
Sundararajan Srikanth, John Ambrose
Publicación
Revista Argentina de Cardioangiología intervencionista
Editor
Colegio Argentino de Cardioangiólogos Intervencionistas
Fecha de publicación
2012-03-30
Registro de propiedad intelectual
© Colegio Argentino de Cardioangiólogos Intervencionistas






Reciba la revista gratis en su correo

Suscribase gratis a nuestra revista y recibala en su correo antes de su publicacion impresa.